Article Text

Statistics from Altmetric.com
Description
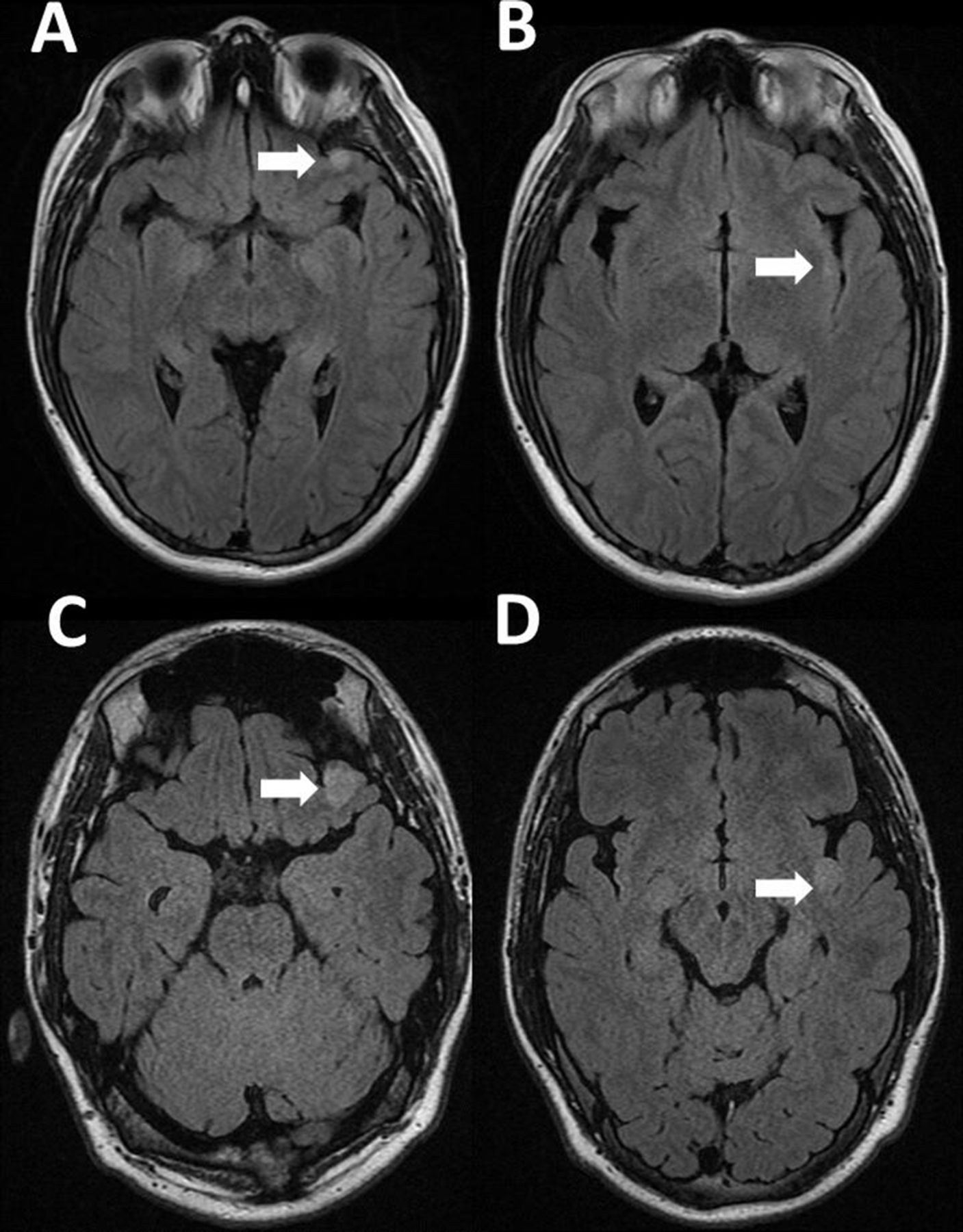
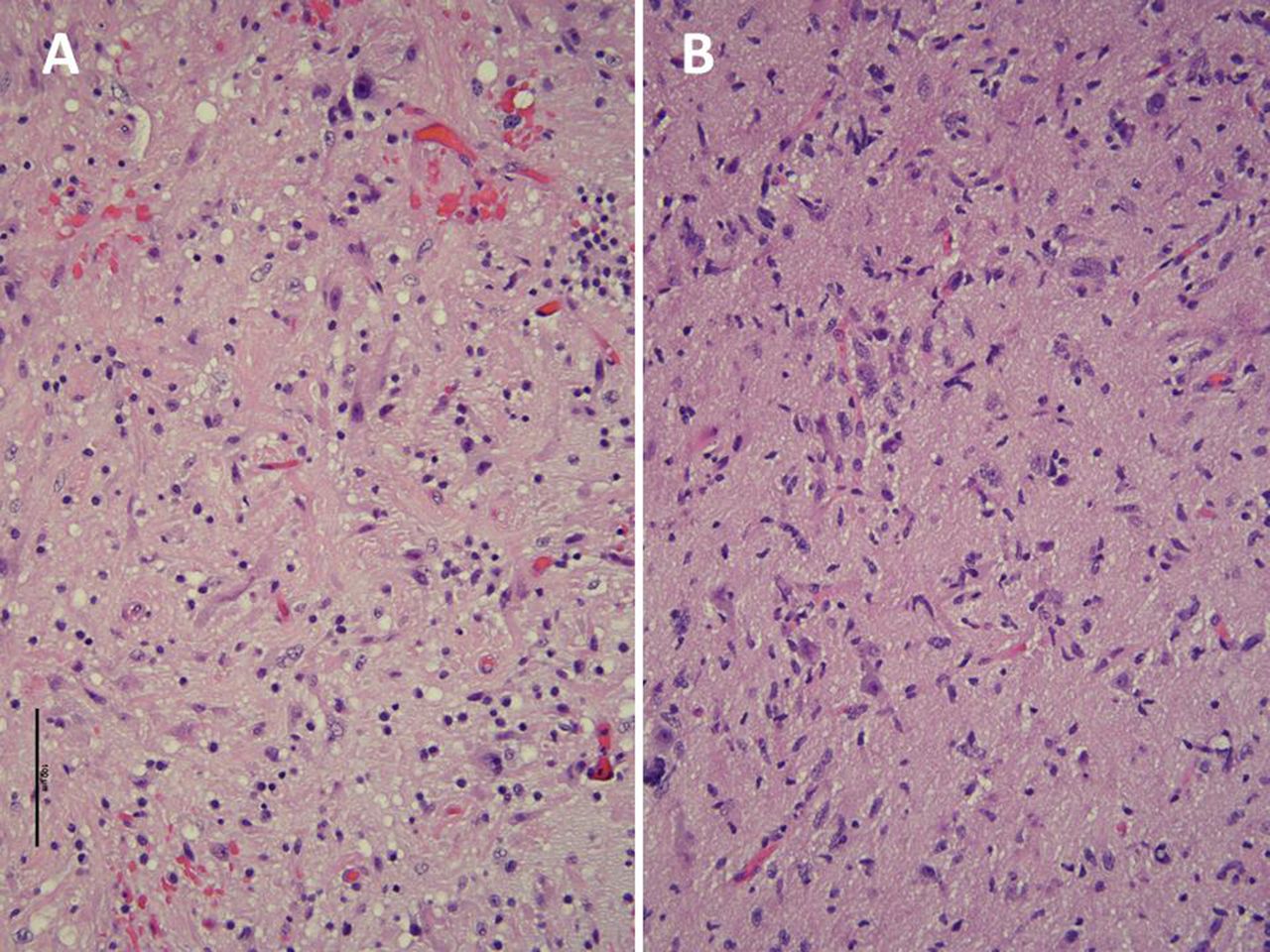
A teenage boy with a history of neurofibromatosis type 1 (NF1) diagnosed in the first year of life presented with focal onset seizures. Neurological examination was normal. MRI of the brain revealed a fluid-attenuated inversion recovery (FLAIR) hyperintense lesion in the left inferior orbital gyrus (figure 1A) and a very subtle hyperintense FLAIR lesion in the left posterior insula (figure 1B), which were monitored by serial neuroimaging. The differential diagnosis at that time included focal areas of signal hyperintensity associated with NF1 versus a low-grade glioma. Four years later MRI demonstrated an enlargement of both lesions in association with recently diagnosed intractable epilepsy (figure 1C–D). Only the frontal lesion showed partial contrast enhancement at presentation and progression (not shown). The patient underwent epilepsy surgery of the dual lesions following a seizure localisation confirmatory testing that included a preoperative videoelectroencephalography and intraoperative electrophysiological monitoring. Neuropathology of the frontal lobe lesion (figure 2A) demonstrated a moderately cellular proliferation of mixed dysplastic neuronal/ganglionic and astrocytic glial cells, with eosinophilic granular bodies and focal lymphocytic infiltrates. Immunohistochemistry demonstrated diffusively positive glial fibrillar acidic protein (GFAP) staining and negative CD34 staining. Antigen KI-67 (Ki-67) had a focal positivity of 5% consistent with a diagnosis of a ganglioglioma with histologic features of a higher-grade tumour. Neuropathology of the non-enhancing insular lesion (figure 2B) revealed a moderately cellular proliferation of elongated glial cells infiltrating around mildly atypical neurons, diffusively positive GFAP staining and a Ki-67 focal positivity of 1%, consistent with a diagnosis of a diffuse astrocytoma. In both tumours, next-generation sequencing was significant for inactivating germline mutations of NF1 and ATM, as well as variants of unknown significance (VUS) in TPR, PIK3R1, PDGFRB and RUNX1. However, the frontal lobe ganglioglioma showed a pathogenic mutation in SMAD4 and a VUS in ATP2B3 while the insular diffuse astrocytoma revealed a VUS in FGFR. Additionally, microarray analysis revealed markedly different chromosomal alterations with hypodiploidy in only the frontal ganglioglioma, suggesting an association with higher-risk disease. More than 3 years following epilepsy surgery, the patient was observed with no progression of either tumour and well-controlled seizures on monotherapy.

Neuroimaging findings of two distinct tumours in a patient with NF1. MRI of the brain revealed a fluid-attenuated inversion recovery (FLAIR) hyperintense lesion in the left inferior orbital gyrus (A) (arrow) and a very subtle hyperintense FLAIR lesion in the left posterior insula (B) (arrow), which were subsequently monitored by serial neuroimaging. Four years later, MRI demonstrated an enlargement of both the orbital gyrus (C) and insular (D) lesion in association with recently diagnosed intractable epilepsy. Only the orbital gyrus lesion showed contrast enhancement at diagnosis and progression (not shown). NF1, neurofibromatosis type 1.

{kind=link}
{kind=link}
Neuropathological features of a ganglioglioma and diffuse astrocytoma in the same patient with NF1. Neuropathology of the frontal lobe lesion demonstrated a moderately cellular proliferation of mixed dysplastic neuronal/ganglionic and astrocytic glial cells, with eosinophilic granular bodies and focal lymphocytic infiltrates (A) Immunohistochemistry demonstrated diffusively positive glial fibrillar acidic protein (GFAP) staining (not shown) and negative CD34 staining (not shown). Antigen KI-67 (Ki-67) had a focal positivity of 5% (not shown) consistent with a diagnosis of a ganglioglioma with histological features of a higher-grade tumour. Neuropathology of the non-enhancing insular lesion. (B) Revealed a moderately cellular proliferation of elongated glial cells infiltrating around mildly atypical neurons, diffusively positive GFAP staining (not shown) and a Ki-67 focal positivity of 1% (not shown), consistent with a diagnosis of a diffuse astrocytoma. NF1, neurofibromatosis type 1.
NF1 is an autosomal dominant condition occurring in approximately 1 in 2500–3000 individuals worldwide.1 It is associated with a loss-of-function mutation in the NF1 tumour suppressor gene (17.q11.2, 283 kb) that encodes a RAS-GAS protein, neurofibromin, involved in the regulation of cell growth and survival.2 3 Non-neurofibroma tumours associated with NF1 have been observed in 7.2% of patients4 and development of concomitant optic pathway and brainstem gliomas have been reported in up to 40% of NF1 patients.5 While other multiple central nervous system tumours have been rarely reported in NF1,6 7 there are no known reports of genetically distinct glial tumours found simultaneously in patients with NF1. The unique histological and molecular features of the frontal ganglioglioma and insular diffuse astrocytoma in our patient adds to the complexity of tumours associated with NF1.
Learning points
Neurofibromatosis type 1 (NF1) is an autosomal dominant condition associated with a significant predisposition to develop central nervous system low-grade glioma that may be associated with epilepsy.
The occurrence of multiple central nervous system tumours in association with NF1 have rarely been reported.
We report the first case of a simultaneous ganglioglioma and diffuse astrocytoma with NF1 and ATM mutations, adding to the complexity of tumours associated with NF1.
Ethics statements
Patient consent for publication
Footnotes
Contributors YK was responsible for the design and writing of the case report. DM was responsive for the design and writing of the case report. ML was responsive for the design and writing of the case report. JRC was responsive for the design and writing of the case report.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Case reports provide a valuable learning resource for the scientific community and can indicate areas of interest for future research. They should not be used in isolation to guide treatment choices or public health policy.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.